Recombinant drug coagulation factor with extended half-life prevents bleeding in children with severe hemophilia A
- Recombinant drug Efanesoctocog alfa (Altuviiio), a genetically engineered factor VIII concentrate with an extended half-life, almost completely prevented spontaneous bleeding among children suffering from hemophilia A, according to a recent study.
- Among 73 patients treated for one year, 65 (88%) remained free of spontaneous bleeding and 47 (64%) had no bleeding episodes of any kind. Moreover, ninety-five percent of the breakthrough episodes resolved after additional injection of the new drug. No treatment-related serious adverse events were reported and none of the patients developed factor VIII inhibitors.
- Unlike older FVIII preparations that require infusions every 2 or 3 days, efanesoctocog alfa can be administered once per week, helping hemophilia patients to live normal lives. The drug is now approved by the FDA and EMA for both treatment and prophylaxis.
Hemophilia A is a genetic disorder characterized by impaired coagulation and frequent bleeding episodes due to a deficiency of factor VIII (FVIII), a crucial step in the blood clotting cascade. The standard treatment approach involves regular infusions of FVIII concentrates, usually every 2-3 days, which significantly burden patients and reduce their quality of life. Introduction of recombinant drug preparations with prolonged duration of action (extended half-life clotting factor concentrates) has partially alleviated this issue, but until recently, the maximum half-life that could be achieved for FVIII products was ~19 hours, which for patients translates into twice weekly administration. This pharmacokinetic barrier is caused by so-called “ceiling effect”, imposed by the coupling of FVIII with von Willebrand factor (VWF), which protects the former from premature degradation. Unfortunately, this protection lasts for no more than 19 hours, setting a limit on the half-life of standard FVIII.
Thankfully, the ceiling effect has been recently overcome with the introduction of genetically engineered factor VIII concentrate, efanesoctocog alfa (Altuviiio). Owing to the sophisticated structural modifications, the half-life of FVIII could be extended to 37-41 hours, which is ~2.5 times higher than for the previous products. Last year, this new drug was shown to be highly effective at preventing bleeding in adult patients, reducing the mean annualized rate from 2.96 on standard FVIII concentrate to 0.69 after initiating efanesoctocog alfa. Now a similar investigation has been conducted in children.
In this pediatric study, published recently in New England Journal of Medicine, 74 patients aged 12 years or younger initiated once-weekly prophylaxis with Altuviiio. The new therapy proved to be highly effective, especially considering that the enrolled children had severe hemophilia with frequent bleeding episodes despite standard prophylactic treatment. The median and mean annualized bleeding rates on the drug were 0.00 and 0.61, respectively. Sixty-five patients (88%) had no spontaneous bleeding and forty-seven patients (64%) remained completely free of bleeding episodes throughout the 52-week follow-up. Bleeding into joints, a painful issue commonly experienced by hemophiliacs, was eliminated in 61 of the treated children (82%). Importantly, there were no serious adverse events related to the drug administration, and no patient developed factor VIII inhibitors, which can render the replacement therapy ineffective and increase the morbidity risk.
In addition to prophylaxis, Altuviiio can also be used for treating the cases of “breakthrough” bleeding, which may occur rarely despite the continuous FVIII administration or more often with on-demand use. In the aforementioned trials, nearly all such cases could be resolved upon a single extra injection of the drug (97% in adults and 95% in children). Efanesoctocog alfa is a fusion protein with a highly complicated structure. It consists of B-domain deleted factor VIII fused to proprietary XTEN polypeptide (developed by a company Amunix Pharmaceuticals) and dimeric Fc, covalently bound to the FVIII-binding domain of von Willebrand factor and two XTEN polypeptide. For clarity, the drug’s structure is illustrated below.
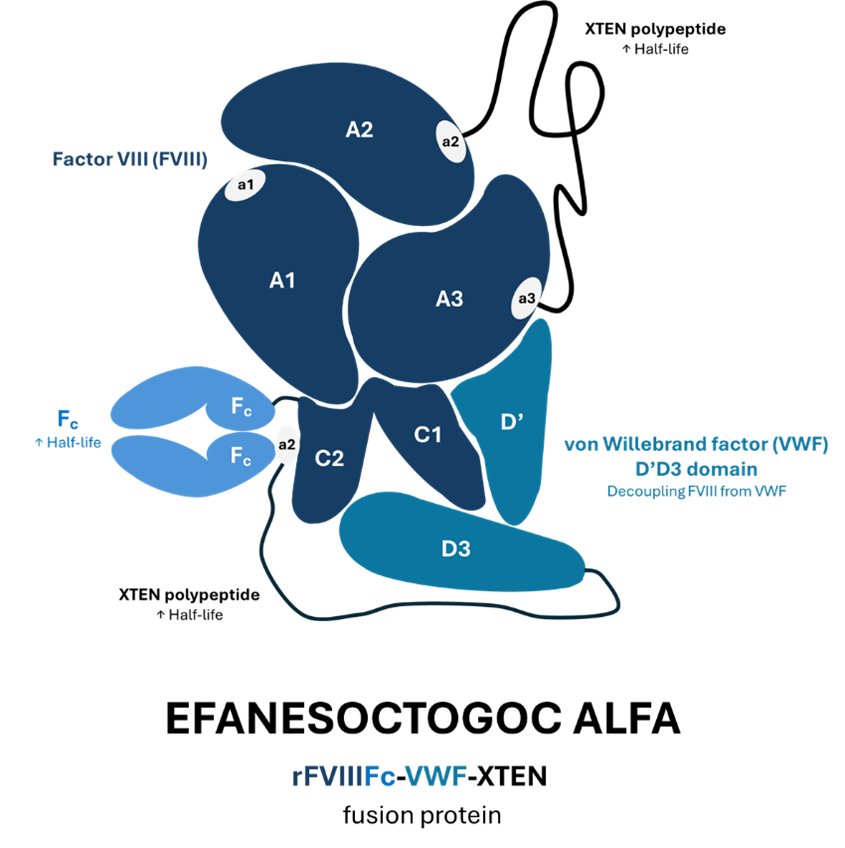
Efanesoctocog alfa has already received approval from the FDA and EMA for the treatment of adults and children with hemophilia A (in February 2023 and April 2024, respectively). However, the newly published trial supplements our knowledge on its efficacy and safety in pediatric population. Altuviiio prevents bleeding episodes regardless of age, causing no major side effects and eliminating the need for up to four weekly infusions of FVIII concentrates.
Prepared by:
Sources and further reading
- Von Drygalski, Annette, et al. “Efanesoctocog alfa prophylaxis for patients with severe hemophilia A.” New England Journal of Medicine 388.4 (2023): 310-318.
- Malec, Lynn, et al. “Efanesoctocog Alfa Prophylaxis for Children with Severe Hemophilia A.” New England Journal of Medicine 391.3 (2024): 235-246.
- Food and Drug Administration (FDA) Altuviiio. Link: https://www.fda.gov/vaccines-blood-biologics/altuviiio.
- European Medicines Agency (EMA). Meeting highlights from the Committee for Medicinal Products for Human Use (CHMP) 22-25 April 2024 (April 26, 2024). Link: https://www.ema.europa.eu/en/news/meeting-highlights-committee-medicinal-products-human-use-chmp-22-25-april-2024.
- https://www.levelsmatterhcp.com/the-vwf-factor-viii-relationship.